Síndrome de Ehlers-Danlos é a designação atribuída a uma doença congénita pouco comum e de difícil diagnóstico que se caracteriza pelo aumento da mobilidade articular e da elasticidade da pele (tecidos conjuntivos). Esta mobilidade anormal tem diferentes implicações no organismo, implicações cuja severidade varia desde a “simples” mobilidade articular e cutânea até a hipermobilidade grave com escoliose, problemas cardíacos e rompimento do baço.
Deve o seu nome a Ehlers que descreveu a hipermobilidade articular em 1901 e a Danlos que por sua vez descreveu a fragilidade cutânea em 1908.
Incidência, Etiologia e Diagnóstico:
Globalmente a incidência é de 1 em 5000 nados-vivos, sendo os tipos I e II os mais frequentes. A hereditariedade destas formas é autossómica.
Face às manifestações comuns às várias formas, é provável que como causa subjacente estejam alterações do mesmo gene. As características já referidas não só caracterizam outras formas mais graves desta síndrome, mas também estão presentes num número elevado de outras doenças. Como tal, o diagnóstico deve ser baseado em dados clínicos mais específicos.
Dois sinais clínicos úteis no diagnóstico da síndrome de Ehlers-Danlos são o sinal de Meténier (eversão fácil das pálpebras superiores) e o sinal de Gorlin (capacidade de tocar no nariz com a língua), ambos denotando uma anormal mobilidade cutânea.
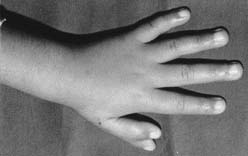
A hipermobilidade articular permite dobrar passivamente o punho e polegar até ao antebraço, assim como, chegar ao umbigo com o braço por trás das costas.
A maioria das crianças com esta síndrome é identificada na infância pela laxidão articular e a hipotonia muscular. Outros sinais associados são a formação fácil de equimoses e a presença de cicatrizes devido à fragilidade cutânea.
Evolução:
A prematuridade é uma das complicações conhecidas. No recém-nascido torna-se evidente a laxidez articular e os lactentes têm atraso do desenvolvimento motor pelo mesmo motivo. Mais tarde os principais problemas envolvem os olhos, a pele, o coração e os ossos.
As escleróticas podem ter um tom azulado. As anomalias oculares manifestam-se em córneas pequenas, estrabismo e alterações da retina. A morfologia dentária pode estar alterada pelas anomalias do esmalte e da dentina. As gengivas são frágeis e a periodontite pode surgir precocemente.
A nível da pele são evidentes a hiperelasticidade cutânea, dificuldades de cicatrização na sequência de pequenas feridas, a formação de cicatrizes finas e pigmentadas e a presença de nódulos subcutâneos em 30% dos doentes. A pele fica com um tom azulado no tempo mais frio e nos adultos são mais evidentes as varizes. Os problemas ósseos mais frequentes são a escoliose, as pernas em arco, pé plano e os pés botos. As hérnias inguinais e umbilicais são também mais frequentes. Ocasionalmente pode haver infecções urinárias (por refluxo vesico-ureteral) e a diminuição da sensibilidade nas extremidades (neuropatia periférica).
Outros riscos associados são as anomalias cardíacas, nomeadamente o prolapso da válvula mitral, a estenose pulmonar e os defeitos septais.
As mulheres afectadas têm um maior risco de hemorragia pós-parto, prolapso do útero ou da bexiga.
Tratamento e Prevenção das Complicações:
O parto de uma grávida com o síndrome de Ehlers-Danlos I-III deve ser planeado adequadamente, visto poder correr o risco de hemorragia e lacerações graves na sequência de uma episiotomia. Neste contexto, é também mais frequente o prolapso rectal e vesical.
Sendo a hereditariedade autossómica dominante, o risco do feto ser afectado é de 50%, o que acarreta uma maior probabilidade de parto prematuro. É também necessário realizar um exame cuidado e pormenorizado do recém-nascido. A elasticidade cutânea e a laxidez articular exagerados (característicos da doença) sugerem este diagnóstico. As avaliações oftalmológicas e cardíacas podem contribuir para esclarecer o diagnóstico e devem ser realizadas periodicamente nos casos confirmados. A fragilidade das gengivas e a hipotonia podem levar a dificuldades alimentares. Em todas as consultas deve ser avaliada a nutrição, crescimento, olhos, coração, pele e articulações. Com o início da dentição e da marcha, estas crianças devem ser orientadas para as respectivas consultas de especialidade.
Na adolescência e na idade adulta, as medidas preventivas devem monitorizar os sistemas já referidos e ser complementadas com os exames considerados necessários pela clínica. Os desportos de contacto devem ser, logicamente, evitados.
Aconselhamento Genético:
O risco de recorrência em futuros filhos de adultos com a síndrome de Ehlers–Danlos tipo I-III é de 50%. A referência a problemas tais como a prematuridade e o prolapso da bexiga e recto na gravidez e parto devem fazer parte do aconselhamento genético. Apesar das várias manifestações desta doença, é importante realçar que a esperança média de vida e inteligência dos indivíduos afectados são consideradas normais. É importante realizar periodicamente avaliações oftalmológicas e cardíacas.
Impacto Social:
Os portadores desta síndrome estão sujeitos a um considerável desgaste emocional pois não só têm que lidar com a doença e todas as consequências físicas desta como sofrem o estigma da “aberração” pois são capazes de movimentos anormais, quebrando os limites de extensão que a maioria dos seres humanos possui.
Note-se que nalgumas feiras dos séculos XIX várias das “aberrações” publicitadas eram na verdade resultado de doenças raras. O homem borracha (síndrome de Ehler-Danlos), a mulher barbuda (hipertricose ou hirsutismo) são exemplos de pessoas doentes que foram exploradas devido à sua condição. É claro que nas actuais sociedades desenvolvidas estes exemplos extremistas não se verificam, no entanto o mesmo tipo de discriminação (talvez mais dissimulada) poderá ser vivenciada pelos portadores pois é um comportamento inato do ser humano (ainda que altame te reprovável) recear tudo o que foge aos padrões tidos como comuns. Deve, por isso, ter-se em atenção possíveis patologias psicológicas que o portador possa vir a desenvolver em consequência do que foi referido.
REFERÊNCIA BIBLIOGRÁFICA:
http://apeslfb.wordpress.com/sindrome-de-ehlers-danlos
POSTADO POR: BIANCA ALMEIDA CRUZ.