
Townes e Brocks, em 1972, descreveram uma síndrome genética de herança autossômica dominante que se caracterizava principalmente por imperfuração anal, dedo polegar com três falanges, outras anormalidades dos dedos do pé ou da mão, sendo relatada também disacusia neurossensorial. Estes autores, cuja síndrome levou seus nomes, relataram que a síndrome poderia ocorrer em razão de mutação genética simples, apresentando expressividade variável.
A Síndrome de Townes-Brocks, devido a sua expressividade variável, pode manifestar-se com vários sinais, dentre eles: anormalidades das mãos, incluindo bifidez do polegar, terceira falange em polegar e polidactilia pré-axial; malformações anais como imperfuração e estenose; anormalidades do trato urinário, incluindo hipospádia, bolsa escrotal bífida e hipoplasia renal; anormalidades do ouvido externo, médio e interno. As anormalidades do ouvido externo incluem microtia, "tags” e "pits" pré-auriculares, orelha com deformidade na região superior da hélix, sendo que nem todos os pacientes apresentarão alguma destas alterações. As alterações do ouvido médio foram descritas em um paciente com Síndrome de Townes-Brocks, como hipoplasia da cabeça do martelo, dismorfismo dos processos longos e curtos da bigorna e anormalidades do formato da janela oval. As alterações estruturais do ouvido interno não são descritas, porém a disacusia neurosensorial é bem conhecida, sendo descrita já por Townes e Brocks.
Este estudo apresenta o caso de um paciente com Síndrome de Townes-Brocks, sem história prévia da patologia em sua família, acreditando tratar-se de mutação nova. O paciente é portador de anormalidade do polegar, dos ouvidos externo e médio, conforme mostrou estudo tomográfico dos ossos temporais.
RELATO DE CASO
Paciente do sexo masculino, 05 anos, nascido de pais sadios consangüíneos, sem história de malformação auricular ou de ouvido interno na família. A gestação transcorreu sem intercorrências, com parto normal a termo. Seus pais tiveram mais dois filhos, sendo nosso paciente o segundo na árvore genealógica. O primeiro, do sexo masculino, faleceu aos três meses de idade sem causa iden-tificada, e a criança mais nova, do sexo feminino, 04 anos, apresenta retardo de crescimento neuropsicomotor não decorrente de alteração cromossômica, segundo avaliação genética.
Ao exame físico, o paciente apresenta estenose de conduto auditivo externo bilateral, mais acentuada à esquerda, associado a discreta deformidade da parte superior da hélix (e à duplicação do polegar da mão esquerda Seu peso e perímetro cefálico estão abaixo do 3º percentil da distribuição normal para a idade, com desenvolvimento somático e intelectual normais. À investigação clínica, não se observou presença de outras malformações.
Na avaliação complementar do ouvido, foi realizada audiometria de tronco cerebral e tomografia computadorizada dos ossos temporais. A audiometria do tronco cerebral revelou latência de todas as ondas aumentadas, mantendo os intervalos entre picos, com limiar de 65 dB HL bilateralmente. O exame tomográfico mostrou mastóide esclerótica bilateralmente, velamento parcial das células da mastóide esquerda e ossículos do ouvido médio sem definição, dando a impressão de bloco único bilateral.
Este estudo apresenta o caso de um paciente com Síndrome de Townes-Brocks, sem história prévia da patologia em sua família, acreditando tratar-se de mutação nova. O paciente é portador de anormalidade do polegar, dos ouvidos externo e médio, conforme mostrou estudo tomográfico dos ossos temporais.
RELATO DE CASO
Paciente do sexo masculino, 05 anos, nascido de pais sadios consangüíneos, sem história de malformação auricular ou de ouvido interno na família. A gestação transcorreu sem intercorrências, com parto normal a termo. Seus pais tiveram mais dois filhos, sendo nosso paciente o segundo na árvore genealógica. O primeiro, do sexo masculino, faleceu aos três meses de idade sem causa iden-tificada, e a criança mais nova, do sexo feminino, 04 anos, apresenta retardo de crescimento neuropsicomotor não decorrente de alteração cromossômica, segundo avaliação genética.
Ao exame físico, o paciente apresenta estenose de conduto auditivo externo bilateral, mais acentuada à esquerda, associado a discreta deformidade da parte superior da hélix (e à duplicação do polegar da mão esquerda Seu peso e perímetro cefálico estão abaixo do 3º percentil da distribuição normal para a idade, com desenvolvimento somático e intelectual normais. À investigação clínica, não se observou presença de outras malformações.
Na avaliação complementar do ouvido, foi realizada audiometria de tronco cerebral e tomografia computadorizada dos ossos temporais. A audiometria do tronco cerebral revelou latência de todas as ondas aumentadas, mantendo os intervalos entre picos, com limiar de 65 dB HL bilateralmente. O exame tomográfico mostrou mastóide esclerótica bilateralmente, velamento parcial das células da mastóide esquerda e ossículos do ouvido médio sem definição, dando a impressão de bloco único bilateral.
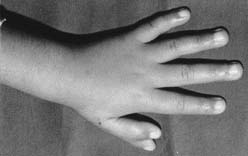
Fotografia mostrando duplicação do polegar da mão esquerda.
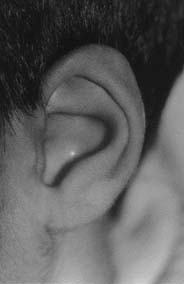
Fotografia mostrando discreta alteração do ouvido externo na região superior da hélix.
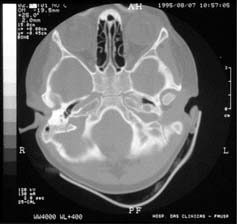
Tomografia computadorizada, em corte axial, revelando estenose de conduto auditivo externo bilateral.
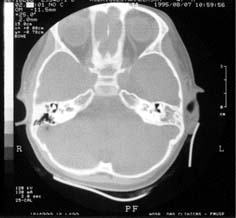
Tomografia computadorizada, em corte axial, onde se observam mastóides com características escleróticas e velamento das células, totalmente à esquerda e parcialmente à direita.
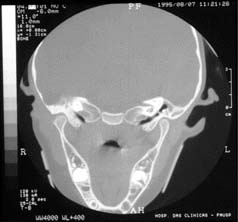
Tomografia computadorizada, em corte coronal, revelando cadeia ossicular mal definida à esquerda.
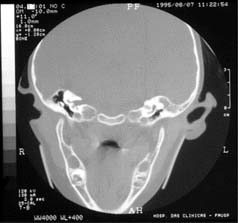
Tomografia computadorizada, em corte coronal, revelando cadeia ossicular mal definida à direita.
DISCUSSÃO
A Síndrome de Townes-Brocks constitui entidade bastante rara, estando descritos aproximadamente 51 casos na literatura, oriundos de vários países como EUA, Austrália, Bélgica, Brasil e Portugal. A transmissão é genética, com provável padrão autossômico dominante e penetrância variável. As malformações anorretais são as mais freqüentes, sendo encontradas em 94% dos casos; as malformações da orelha externa, em 74% dos casos, e disacusia neurosensorial, em 25% destes6. Nos casos onde o ouvido é afetado, o acometimento pode ser uni ou bilateral.
A associação entre qualquer manifestação do complexo anotia/microtia e polidactilia pré-axial indica, com alta probabilidade, o diagnostico de Síndrome de Townes-Brocks, de herança autossômica dominante, apesar da ausência de atresia/defeito de implantação anal. A alteração cromossômica deve ter aparecido no paciente como resultado de mutação nova, não tendo aparentemente relação com a consangüinidade dos pais.
A perda auditiva em pacientes com Síndrome de Townes-Brocks foi revelada no artigo de descrição da síndrome pelos autores que lhe conferiram o nome, como sendo quadro de disacusia sensorioneural. Estes autores mostraram uma família onde seis membros foram acometidos pela síndrome, sendo realizado teste auditivo em somente quatro membros, que revelou perda auditiva sensorioneural, de grau leve a moderado, em todos os exames. Rossmiller, em 1994, publicou artigo onde uma família que apresentava seis indivíduos acometidos pela síndrome foi submetida a investigação audiológica. Esta avaliação mostrou alterações auditivas envolvendo tanto o componente sensorioneural como condutivo, ou seja, dissacusia mista em todos os pacientes. A otoscopia não mostrou indícios de patologia de ouvido médio, e a timpanometria em somente um indivíduo apresentou grande amplitude, sugerindo descontinuidade ossicular. A discriminação auditiva variou de 64% a 96%. A perda auditiva era significativamente pior nas freqüências agudas em relação às freqüências mais graves, havendo melhor limiar auditivo nos pacientes mais jovens, quando comparados aos mais idosos. Desta forma, além da verificação da disacusia mista nos pacientes com Síndrome de Townes-Brocks, foi sugerido por Rossmiller perda auditiva progressiva nestes pacientes.
A função anormal do ouvido médio nestes pacientes é sugerida pela perda auditiva condutiva verificada no estudo audiológico. Descrição detalhada das alterações da cadeia ossicular do ouvido médio depende de timpanotomia exploradora ou de exame pós-mórtem dos ossos temporais dos pacientes acometidos pela síndrome. Por este motivo, tem-se poucos dados na literatura referentes às alterações anatômicas da cadeia ossicular. As alterações do ouvido médio foram descritas por Ferraz (1989) em um paciente com Síndrome de Townes-Brocks, como hipoplasia da cabeça do martelo, dismorfismo dos processos longos e curtos da bigorna e anormalidades do formato da janela oval.
Concluimos, então, que a perda auditiva dos pacientes com Síndrome de Townes-Brocks é congênita, hereditária, progressiva e caracterizada por transmissão autossômica dominante. A deficiência auditiva pode ser neurosensorial, condutiva ou mista, com severidade variável. As deformidades dos ouvidos externo e médio apresentam penetrância incompleta, nào sendo relatadas em todos os pacientes.
A Síndrome de Townes-Brocks constitui entidade bastante rara, estando descritos aproximadamente 51 casos na literatura, oriundos de vários países como EUA, Austrália, Bélgica, Brasil e Portugal. A transmissão é genética, com provável padrão autossômico dominante e penetrância variável. As malformações anorretais são as mais freqüentes, sendo encontradas em 94% dos casos; as malformações da orelha externa, em 74% dos casos, e disacusia neurosensorial, em 25% destes6. Nos casos onde o ouvido é afetado, o acometimento pode ser uni ou bilateral.
A associação entre qualquer manifestação do complexo anotia/microtia e polidactilia pré-axial indica, com alta probabilidade, o diagnostico de Síndrome de Townes-Brocks, de herança autossômica dominante, apesar da ausência de atresia/defeito de implantação anal. A alteração cromossômica deve ter aparecido no paciente como resultado de mutação nova, não tendo aparentemente relação com a consangüinidade dos pais.
A perda auditiva em pacientes com Síndrome de Townes-Brocks foi revelada no artigo de descrição da síndrome pelos autores que lhe conferiram o nome, como sendo quadro de disacusia sensorioneural. Estes autores mostraram uma família onde seis membros foram acometidos pela síndrome, sendo realizado teste auditivo em somente quatro membros, que revelou perda auditiva sensorioneural, de grau leve a moderado, em todos os exames. Rossmiller, em 1994, publicou artigo onde uma família que apresentava seis indivíduos acometidos pela síndrome foi submetida a investigação audiológica. Esta avaliação mostrou alterações auditivas envolvendo tanto o componente sensorioneural como condutivo, ou seja, dissacusia mista em todos os pacientes. A otoscopia não mostrou indícios de patologia de ouvido médio, e a timpanometria em somente um indivíduo apresentou grande amplitude, sugerindo descontinuidade ossicular. A discriminação auditiva variou de 64% a 96%. A perda auditiva era significativamente pior nas freqüências agudas em relação às freqüências mais graves, havendo melhor limiar auditivo nos pacientes mais jovens, quando comparados aos mais idosos. Desta forma, além da verificação da disacusia mista nos pacientes com Síndrome de Townes-Brocks, foi sugerido por Rossmiller perda auditiva progressiva nestes pacientes.
A função anormal do ouvido médio nestes pacientes é sugerida pela perda auditiva condutiva verificada no estudo audiológico. Descrição detalhada das alterações da cadeia ossicular do ouvido médio depende de timpanotomia exploradora ou de exame pós-mórtem dos ossos temporais dos pacientes acometidos pela síndrome. Por este motivo, tem-se poucos dados na literatura referentes às alterações anatômicas da cadeia ossicular. As alterações do ouvido médio foram descritas por Ferraz (1989) em um paciente com Síndrome de Townes-Brocks, como hipoplasia da cabeça do martelo, dismorfismo dos processos longos e curtos da bigorna e anormalidades do formato da janela oval.
Concluimos, então, que a perda auditiva dos pacientes com Síndrome de Townes-Brocks é congênita, hereditária, progressiva e caracterizada por transmissão autossômica dominante. A deficiência auditiva pode ser neurosensorial, condutiva ou mista, com severidade variável. As deformidades dos ouvidos externo e médio apresentam penetrância incompleta, nào sendo relatadas em todos os pacientes.
REFERÊNCIA BIBLIOGRÁFICA:
http://www.arquivosdeorl.org.br/conteudo/acervo_port.asp?id=15
Nenhum comentário:
Postar um comentário