
As Atrofias Musculares Espinhais (AME) têm origem genética e caracterizam-se pela atrofia muscular secundária à degeneração de neurônios motores localizados no corno anterior da medula espinhal. A AME, a segunda maior desordem autossômica recessiva fatal, depois da Fibrose Cística (1:6000), afeta aproximadamente 1 em 10.000 nascimentos , com uma freqüência de doentes de 1 em 50 portadores. Casais que tiveram uma criança afetada têm 25% de risco de recorrência em cada gravidez subseqüente.
Os caracteres autossômicos recessivos são transmitidos por ambos os progenitores. Nos casamentos consangüíneos, há uma probabilidade maior de nascerem filhos com caráter recessivo, pois indivíduos aparentados têm maior probabilidade do que os não parentes de serem heterozigotos para o mesmo gene mutante. Desta forma, há uma proporção maior de casamentos consangüíneos entre os progenitores de afetados por um caráter recessivo, do que entre progenitores de pessoas normais.
Hipotonia, paralisia, arreflexia, amiotrofia e miofasciculação constituem os sinais definidores das AMEs, doenças autossômicas recessivas ligadas ao cromossoma 5, relacionadas ao gene SMN. São subdivididas em três grupos de acordo com a idade de início e evoluem tão mais rapidamente quanto mais cedo começam.
O grupo I (doença de Werdnig-Hoffmann) pode se manifestar desde a fase intra-útero até os 3 meses de vida extra-uterina. Durante a gravidez verifica-se pouca movimentação fetal e o recém-nascido apresenta fraqueza acentuada das musculaturas proximal e intercostal. A postura é bem diferente daquela esperada para um neonato sadio: as pernas estão estendidas, em abdução e rotação externa, os membros superiores ficam largados (postura em batráquio). O tórax é estreito e geralmente há pectum excavatum. Tem dificuldades de sucção, de deglutição e respiratórias. As infecções respiratórias se repetem, sendo a causa do óbito na maioria dos casos.
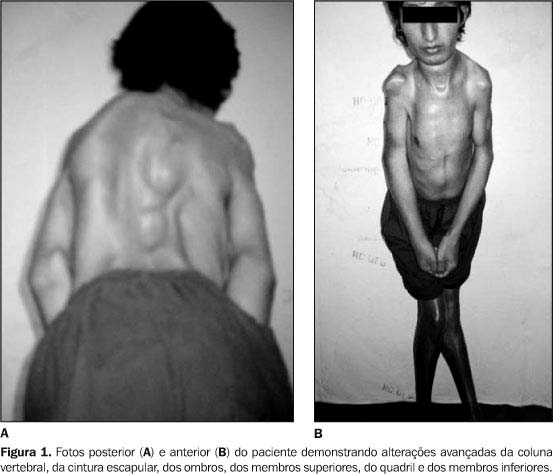
Nos grupos II e III os sintomas se manifestam entre 3 e 15 meses e de 2 anos a vida adulta, respectivamente. O quadro clínico caracteriza-se por deterioração motora após um período de desenvolvimento aparentemente normal. Na forma tipo II, ou forma intermediária, a criança adquire a habilidade de sentar mas tem uma parada do desenvolvimento motor a partir deste marco. A forma tipo III ou forma juvenil ou doença de Wolfhart-Kugelberg-Welander começa normalmente a dar os primeiros sinais de fraqueza depois de 1 ano de idade ou até mais tarde. De maneira lenta a fraqueza nas pernas faz com que as crianças caiam mais, tenham dificuldade para correr, subir escadas e levantar do chão. Também aparece fraqueza nos ombros, braços e pescoço. Às vezes a doença é confundida com Distrofia Muscular. A fraqueza aumenta com o passar dos anos e a cadeira de rodas se torna necessária em algum momento na vida adulta. Os sinais da doença são menos proeminentes e a progressão mais lenta, sendo que deformidades ósteo-articulares, principalmente escoliose, aparecem à medida que o curso se prolonga. Há outras formas, variantes da AME, sem um padrão perfeitamente definido de herança, de comportamento clínico heterogêneo eventualmente associado a outras manifestações.
Todas as três formas acima não têm cura definitiva. No entanto a fisioterapia, os bons cuidados no acompanhamento clínico e alguns aparelhos ortopédicos ajudam a manter a independência destas crianças, a função de seus músculos e a integridade física e mental.
O pediatra tem uma importância fundamental. Cabe a ele reconhecer que uma criança está com problemas, ou por ser hipotônica, ou por ter pouca movimentação espontânea, ou por estar atrasada em algum marco do desenvolvimento. Quanto mais cedo o reconhecimento, mais precoce será o encaminhamento ao especialista certo, mais rápido o diagnóstico e mais eficaz a terapia de suporte que podemos oferecer.
A realização do diagnóstico não deve afastar o pediatra de seu paciente. Mais do que nunca a paciente e sua família precisam de um apoio para as eventuais complicações clínicas associadas a esta doença. Além disto, a criança continua sendo criança, com as necessidades de puericultura inerentes a sua faixa etária.
As doenças neuromusculares, caracterizam-se por situações decorrentes de problemas localizados na ponta anterior da medula, nos nervos periféricos, nas placas mioneurais ou nos músculos. As atrofias Musculares Espinhais e as Distrofias Musculares são exemplos de doenças neuromusculares de origem genética.
REFERÊNCIA BIBLIOGRÁFICA:
http://www.atrofiaespinhal.org/oque.php
POSTADOR POR:
STEFÂNIA DE JESUS PEREIRA

