A Hemofilia é um distúrbio de coagulação sangüínea geneticamente determinada, sendo três tipos mais freqüentes dessas coagulopatias, a hemofilia tipo A, B e a doença de Von Willebrand (RODRÍGUEZMERCHÁN, 1997), emboras as outras deficiências de coagulação hereditárias menos freqüentes possam, às vezes, apresentar quadro hemorrágico severo.
Historicamente, a mortalidade dos portadores de hemofilia e outros distúrbios congênitos de coagulação era devida à hemorragia incontrolada associada à severa deficiência de fatores de coagulação VII e IX (DIAMONDSTONE; ALEDORT; GOEDERT, 2002).
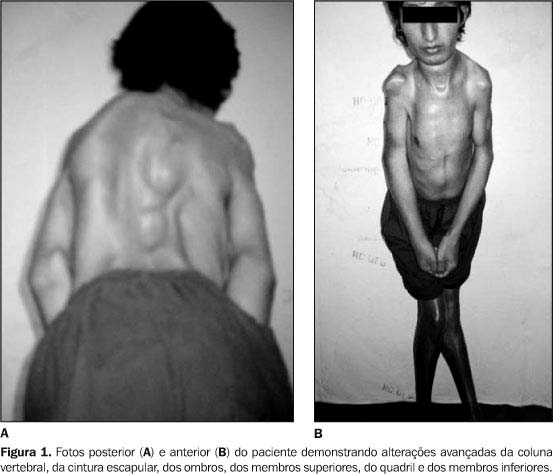
Sem a intervenção médica através da reposição dos fatores de coagulação e o acompanhamento fisioterapêutico preventivo, com a finalidade de se evitar incapacidade funcional e proporcionar melhor qualidade de vida para esses indivíduos, torna-se muito difícil evitar as complicações músculo esqueléticas dessa moléstia que inclui limitações de movimentos articulares, hemartrose, hemorragias tissulares, aderências articulares fibróticas, alterações de marcha, assimetria de forças musculares, contraturas e artrite hemofílica; alterando a vida dos indivíduos hemofílicos. As hemartroses agudas são os tipos mais freqüentes de derrame no paciente com hemofilia. (RIBBANS; GIANGRANDE; BEETON, 1997).
Por sua determinação genética, os episódios de hemorragias iniciam-se precocemente na vida das crianças, assim sendo, Rodríguez-Merchán (1997) defende uma profilaxia contínua entre 2 a 18 anos para a redução da incidência de sinovite hemofílica crônica e prejuízos articulares, que permanecem como as principais causas de incapacidade para o paciente hemofílico.
A protelação e/ou um tratamento inadequado podem gerar uma série de alterações patológicas dentro da articulação, levando a artropatia dolorosa (RIBBANS; GIANGRANDE; BEETON, 1997). Rev. biociênc.,Taubaté, v.9, n.1, p.37-45, jan-mar 2003.
O cuidado com o paciente hemofílico envolve uma equipe conduzida pelo médico hematologista, mas também inclui a atuação de um ortopedista e um fisioterapeuta, pois ótimos resultados têm sido obtidos pela combinação do fator de reposição, repouso e reabilitação supervisionada associada à crioterapia (RIBBANS; GINAGRANDE; BEETON, 1997). A hemofilia é uma doença cara, pois seu tratamento é extremamente dependente de fatores de reposição para a coagulação (ROGOFF et al., 2002). Porém, a introdução do tratamento com plasma derivado do fator VIII (FVIII) humano tem proporcionado aos pacientes com hemofilia. A uma qualidade e expectativa de vida similar à da população em geral (SUITER, 2002).
É conhecido que os níveis de fator VIII (hemofilia A) aumentam com o stress, exercício físico, uso de epinefrina e na gravidez (MARINHO, 1985). Os benefícios de uma musculatura forte para proteção das articulações foram descritos por Boone (1966) e em 1974, a mesma autora descreveu que programas de exercícios constantes preveniam e reduziam as hemorragias intra-articulares.
Não há uma incidência verdadeira conhecida, porém estima-se que nos EUA aproximadamente, 8 a 11 em 100.000 da população sejam portadores de hemofilia (RODRÍGUEZ-MERCHÁN, 1997), a maioria no sexo masculino.
No Brasil, também não há uma estimativa abrangente sobre a verdadeira incidência da hemofilia, mas sabe-se que há numerosos casos nos serviços públicos e é necessário especializar o acompanhamento fisioterapêutico a esse tipo de paciente. Para isso, é fundamental o estudo dessa doença e suas respostas ao tratamento por meios físicos.
Dentro desse contexto, o objetivo deste trabalho é elucidar o papel do fisioterapeuta na hemofilia, visto que a fisioterapia dispõe de vários agentes físicos que podem ser aplicados de acordo com a fase da doença.
Apenas apresentaremos uma hipótese dos recursos que podem ser usados em cada fase que o hemofílico se encontrar.
TRATAMENTO
O tratamento precoce é necessário para evitar as complicações da doença. O progresso contínuo de o prejuízo articular depois das hemartroses, potencialmente levam a sinovites crônicas, artropatia destrutiva e geralmente à dor.
Raramente realiza-se, ainda na atualidade, procedimento cirúrgico para o tratamento das artropatias hemofílicas severas, mesmo os de baixo risco como a artroscopia. Recomenda-se que esses procedimentos sejam realizados em centros especializados ao atendimento do indivíduo hemofílico (EICKHOFF et al., 1997). Essa recomendação torna ainda mais importante o tratamento preventivo para os casos de hemofilia, somando-se com os dados de grande risco de fracasso obtido nas artroplastias totais de joelho em hemofílicos, que devido ao freqüente desenvolvimento de artropatia dessa articulação necessitam da colocação de prótese articular numa idade muito jovem (NORIAN et al., 2002).
O tratamento conservador com base na reposição de fatores de coagulação é o mais utilizado, apesar de não representar a cura para o indivíduo hemofílico, possibilita o controle de episódios hemorrágicos. Sobretudo, progresso significante tem sido feito recentemente na terapia genética para a hemofilia (VANDENDRIESSCHE; COLLEN; CHUAH, 2001).
Em 2000, realizou-se o primeiro tratamento com sucesso através da terapia genética para indivíduos hemofílicos (HERZOG; HAGSTROM, 2001), porém ainda tem alto custo e não tem abrangência significativa para os portadores dessa coagulopatia, entretanto a transferência genética tornou-se uma potencial e importante possibilidade de tratamento para a hemofilia, ainda em experimentação (MONAHAN; WHITE II, 2002).
ATUAÇÃO DA FISIOTERAPIA
Para o estabelecimento de um plano de tratamento adequado, é necessária uma avaliação fisioterapêutica que inclua entrevista, exame físico e exames de provas funcionais. Na entrevista deverão ser coletados dados a respeito do paciente, dos seus hábitos de vida, sua história pregressa e a história da moléstia atual. O exame físico deve constar de goniometria, perimetria articular, perimetria muscular, provas de função muscular, análise da postura e da coluna vertebral e um exame de comprimento dos membros inferiores (uma vez que os sangramentos repetidos na articulação do joelho provocam estímulos dos centros de crescimento da superfície óssea articular), além de investigação de comprometimentos respiratórios e presença de dor. Também são importantes os exames de provas funcionais, como a análise da marcha (em superfície plana e inclinada), do equilíbrio estático e dinâmico e um teste de resistência, que consiste na prova dos exercícios que integrarão o esquema terapêutico definitivo (RIBEIRO; SOARES, 1986).
A partir da avaliação inicial deverá ser definido um plano de tratamento fisioterapêutico, necessariamente individualizado, considerando a fase do processo hemorrágico, a intensidade da hemorragia, a dor, a idade do indivíduo e o grau de lesão. Em linhas gerais, os objetivos do tratamento serão o controle da dor, a prevenção de deformidades, a prevenção de complicações respiratórias ou vasculares no paciente acamado, a recuperação da capacidade funcional de um músculo ou de uma articulação, a manutenção de um equilíbrio estático e dinâmico do sistema músculo-esquelético, o estímulo à participação da família e a reintegração do indivíduo no seu meio social e profissional (MARIE GAL; NAGATA, 1985).
TERAPIA PROPOSTA
Preventiva
Um programa preventivo deve incluir cinesioterapia progressiva, incentivo à prática de esportes com baixo impacto, principalmente a natação e orientação quanto aos cuidados necessários para evitar as hemorragias (BATTISTELLA et al., 1985). Cabe ainda ao fisioterapeuta esclarecer os familiares a respeito desta moléstia a fim de que possam contribuir de maneira correta no tratamento e evitem comportamentos superprotetores (BATTISTELLA et al.,1985). Deve-se ensinar também aos pais que não puxem o bebê pelos membros superiores para evitar lesões e hemorragias logo nessa primeira fase da vida.
Fase aguda das Hemorragias
· Imobilização: No caso de sangramento articular, imobilizar em goteira de gesso ou de material termoplástico, como o polipropileno, por no máximo quatro dias. Este procedimento é utilizado para o alivio da dor e para a prevenção de deformidades (BATTISTELLA et al., 1985; RIBEIRO; SOARES, 1986; MARIE GAL; NAGATA, 1985).
· Nos hematomas musculares, a imobilização do músculo comprometido deve ser feita através de repouso (MARIE GAL; NAGATA, 1985). No entanto, devido à atrofia muscular gerada, a imobilização deve ser mantida por curtos períodos e somente quando absolutamente necessária, sempre incentivando a realização de contrações isométricas (BATTISTELLA et al.,1985).
· Uso de órteses, com o intuito de preservar o segmento afetado (MARIE GAL; NAGATA, 1985). Algumas vezes, ao invés da goteira gessada, podem ser utilizados suportes para deambulação, com o intuito de evitar sobrecarga na articulação (BATTISTELLA et al., 1985).
· Crioterapia: São utilizados, mesmo durante a imobilização, aplicações de gelo, gel ou gases refrigerantes na forma de spray, durante 20 minutos (ANDREWS, 2000). É indicada para diminuir a dor, através da redução da atividade dos receptores sensitivos da condução nervosa e da excitabilidade das fibras A delta; para auxiliar na contenção das hemorragias, através da lentificação dos processos metabólicos e da proteólise e também para obtenção de relaxamento muscular, através da diminuição da atividade do sistema músculo-fusal responsável pela manutenção do tônus.
· Eletroterapia: Pode ser utilizada para a reabsorção do hematoma, para analgesia ou para estímulo trófico (MARIE GAL; NAGATA, 1985). As correntes contínuas poderão ser utilizadas para realização de iontoforese com a finalidade da reabsorção do hematoma. Devido a sua ação repulsiva, contra-irritativa e vasodilatadora, o bicloridrato de histamina a 1/10.000 é comumente utilizado. No entanto, em muitos casos a histamina é irritativa para a pele. Nestes casos, a simples galvanização é suficiente, uma vez que as propriedades eletroforéticas da corrente representam o maior benefício da iontoforese (MARIE GAL; NAGATA, 1985). As correntes diadinâmicas e as interferenciais também aceleram a reabsorção de fluidos, além de manter o trofismo muscular.
· Cinesioterapia:Deve ser instituída precocemente, respeitando a dor do paciente (BATTISTELLA et al.,1985). Na fase aguda, deverão ser empregadas contrações isométricas. Tais exercícios são importantes por não provocarem dor, enquanto mantêm e desenvolvem força muscular (RIBEIRO; SOARES, 1986; MARIE GAL; NAGATA, 1985). Deverão ser trabalhados os músculos ligados diretamente à articulação comprometida e todos aqueles que fornecem equilíbrio dinâmico e estático de finalidade funcional (MARIEGAL; NAGATA, 1985). É importante o cuidado com a mobilização passiva, que deve até ser evitada para não agravar as lesões.
· É importante lembrar que o repouso do segmento acometido é de vital importância na fase aguda; assim, maiores esforços ou sobrecarga sobre a região afetada devem ser evitados (RIBEIRO; SOARES, 1986).
Emprego de Ultra-Som, no caso de fibrose (MARIE GAL; NAGATA, 1985) Tratamento Postural: Posturas adequadas devem ser mantidas no leito e fora dele (RIBEIRO; SOARES,1986; MARIE GAL; NAGATA, 1985).
· Nas atividades de vida diária devem ser utilizados, sempre que necessário dispositivos de apoio (RIBEIRO; SOARES, 1986).
Fase Crônica
- Calor: Os efeitos fisiológicos a serem obtidos com a termoterapia de adição são a vasodilatação, a liberação de substâncias vasoativas e o relaxamento do espasmo muscular. Calor superficial ou calor profundo podem ser utilizados para facilitar a mobilização articular e aliviar a dor (MARIE GAL; NAGATA, 1985).Cinesioterapia: Os exercícios terapêuticos visando a funcionalidade, a manutenção e o fortalecimento da musculatura, o restabelecimento e a manutenção da amplitude de movimento articular, o relaxamento, o reforço do esquema corporal, a coordenação, o equilíbrio, a orientação postural, e a prevenção de complicações respiratórias e vasculares (MARIE GAL; NAGATA, 1985). A cinesioterapia deve ser necessariamente, progressiva, iniciando com mobilizações ativas assistidas, como por exemplo, mediante o uso de splints ou molas (suspensoterapia) (RIBEIRO; SOARES, 1986). Na seqüência, devem ser orientados os exercícios ativos livres contra a gravidade, oferecendo como resistência ao movimento apenas o peso do segmento corporal a ser trabalhado. Posteriormente, deve ocorrer incremento progressivo na resistência, manualmente ou através de pesos. Assim, com o progredir do quadro clínico do paciente, pode-se fazer uso da bicicleta ergométrica, de outros dispositivos de mecanoterapia ou de técnicas manuais, como o Kabat (RIBEIRO; SOARES 1986; MARIE GAL; NAGATA, 1985).No entanto, os exercícios isotônicos contra-resistidos são parcialmente contraindicados para pacientes com hemofilia, devido ao risco de aparecimento precoce de artrose fêmoro-patelar nesses pacientes. Restrição semelhante deve ser feita aos exercícios passivos, devido ao risco de desencadearem hemorragias articulares e/ou musculares (BATTISTELLA et al., 1985).
- Hidroterapia: A hidroterapia fornece uma proteção ao sistema músculo-esquelético e permite a retomada da cinesioterapia e da marcha desde a fase subaguda. Além disso, com o avançar do tratamento, a água pode ser usada para empregar resistência aos exercícios na fase crônica (RIBEIRO; SOARES, 1986; MARIE GAL; NAGATA, 1985).
- Eletroterapia: Pode ser usada para recuperar o déficit neuromuscular depois de uma neuropatia por compressão (RIBEIRO; SOARES, 1986).
- Crioterapia: Pode ser usada como medida profilática quando ao fim dos exercícios verifica-se um aumento da temperatura na região. No entanto, a crioterapia é pouco utilizada na fase crônica. (RIBEIRO; SOARES, 1986). Orientação Postural: Deve haver uma adequada orientação para que seja adotada uma postura correta nas atividades de vida diária (RIBEIRO; SOARES, 1986).
RESULTADOS
Diante do exposto, os pacientes que se submetem ao tratamento fisioterapêutico adequado precoce evoluem satisfatoriamente, com controle do derrame intra-articular, alívio da dor, manutenção da capacidade funcional das articulações, prevenindo assim deformidades articulares, de acordo com as evoluções dos pacientes hemofílicos que foram observadas.
CONCLUSÃO
Por meio deste trabalho, nota-se que o tratamento fisioterapêutico para o hemofílico é importante à medida que previne e reduz as complicações dessas patologias. Assim, sugerimos um tratamento individualizado precoce para prevenir as complicações como artropatias hemofílicas, as quais se tornam extremamente incapacitantes com a evolução dessa doença hereditária.
REFERÊNCIA BIBLIOGRÁFICA
http://periodicos.unitau.br/ojs-2.2/index.php/biociencias/article/viewFile/73/51
PORTADO POR:
THAISA CHRISTINA DUTRA PEREIRA