
Histórico:
Em 1841, West em uma carta dramática ao editor do "The Lancet", apresentou o problema de seu filho com espasmos em flexão que se repetiam diariamente em ataques de 10 a 20 contrações que levaram a criança a um retardo mental, apesar de todos os tratamentos usados e possíveis para aquela época.
Esta síndrome neurológica foi descrita pela primeira vez em 1949 por Vasquez y Turner para sociedade Argentina de Pediatria, com dez casos de uma "nova síndrome" que apresentavam crises nos lactantes, com alterações específicas no traçado eletroencefalográfico (EEG), estando associadas à deterioração mental, as quais propuseram chamar Epilepsia em Flexão.
Em 1952, os autores Gibbs e Gibbs criaram o termo Hipsarritmia (hypos altura e rhytmos ritmo) para o registro EEG destes pacientes, o que passou a caracterizar a maioria das descrições desta síndrome. Portanto, trata-se de uma entidade eletroclínica caracterizada por espasmos quase sempre em flexão e por um traçado EEG típico denominado hipsarritmia ou disritmia maior lenta. As crises clínicas têm recebido outras denominações: espasmos saudatórios , espasmo infantil, massive jerks, Blitz und NichtKrampf, tic de salaam e pequeno mal propulsivo.
Etiologia
Esta síndrome neurológica foi descrita pela primeira vez em 1949 por Vasquez y Turner para sociedade Argentina de Pediatria, com dez casos de uma "nova síndrome" que apresentavam crises nos lactantes, com alterações específicas no traçado eletroencefalográfico (EEG), estando associadas à deterioração mental, as quais propuseram chamar Epilepsia em Flexão.
Em 1952, os autores Gibbs e Gibbs criaram o termo Hipsarritmia (hypos altura e rhytmos ritmo) para o registro EEG destes pacientes, o que passou a caracterizar a maioria das descrições desta síndrome. Portanto, trata-se de uma entidade eletroclínica caracterizada por espasmos quase sempre em flexão e por um traçado EEG típico denominado hipsarritmia ou disritmia maior lenta. As crises clínicas têm recebido outras denominações: espasmos saudatórios , espasmo infantil, massive jerks, Blitz und NichtKrampf, tic de salaam e pequeno mal propulsivo.
Etiologia
A Síndrome de West pode ser dividida em dois grupos, com relação à causa: o criptogênio (quando a causa é desconhecida), onde o lactente é normal até os inícios dos espasmos, sem qualquer lesão cerebral detectável; e o grupo sintomático (de causa conhecida), onde há prévio desenvolvimento neuropsicomotor anormal , alterações ao exame neurológico e/ou lesões cerebrais identificadas por exames de imagem ( tomografia computadorizada, ressonância magnética, etc). Em 1991, foi proposta a hipótese da existência de uma forma idiopática , com evolução benigna no tratamento em curto prazo.
Em aproximadamente 80% dos casos a síndrome de West é secundária, o que vale dizer que depende de uma lesão cerebral orgânica. Em muitos casos é possível determinar a etiologia da síndrome: encefalites a vírus, anoxia neonatal, traumatismo de parto , toxoplasmose, Síndrome de Aicardi, esclerose tuberosea de Bounerville. Na presença da Síndrome de West, uma exaustiva investigação deve ser feita: TC ou RNM, teste de testagem de erros inatos do metabolismo. Outro tipo de crises, além dos espasmos, pode estar também associado.
Incidência
Em aproximadamente 80% dos casos a síndrome de West é secundária, o que vale dizer que depende de uma lesão cerebral orgânica. Em muitos casos é possível determinar a etiologia da síndrome: encefalites a vírus, anoxia neonatal, traumatismo de parto , toxoplasmose, Síndrome de Aicardi, esclerose tuberosea de Bounerville. Na presença da Síndrome de West, uma exaustiva investigação deve ser feita: TC ou RNM, teste de testagem de erros inatos do metabolismo. Outro tipo de crises, além dos espasmos, pode estar também associado.
Incidência
Inicia-se quase sempre no primeiro ano de vida, principalmente entre os 4 e 7 meses de idade. O sexo masculino é o mais afetado, na proporção de 2 para 1.
Aspectos Eletroencefalográficos
Aspectos Eletroencefalográficos
As características principais e um registro de EEG com hipsarritmia são:
Desorganização marcante e constante da atividade basal;
Elevada amplitude dos potenciais;
Ondas lentas delta de voltagem muito elevada (" ondas em montanhas ");
Períodos (salvas), habitualmente breves, de poliondas e polipontas onda;
Período de atenuação da voltagem que, em alguns casos, parece chegar a silêncio elétrico.
O eletroencefalograma, ao lado do quadro clínico, é fundamental para diagnóstico, recebendo traçado anormal o nome hipsarritmia. O EEG se caracteriza pelo seu aspecto anárquico, com ondas de grande amplitude, lentas e pontas-ondas lentas. Não há ritmo de base organizada.
Quadro Clínico
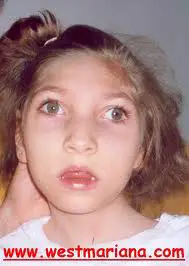
A síndrome de West consiste numa tríade de sinais clínicos e eletroencefalográficos atraso do desenvolvimento, espasmos infantis e traçados eletroencefalográficos com padrão de hipsarritmia. As crises são traduzidas por espasmos ou uma salva de espasmos com seguintes características flexão súbita da cabeça, com abdução dos membros superiores e flexão das pernas (espasmos mioclônico maciço) é comum a emissão de um grito por ocasião do espasmo. Cada crise dura em média alguns segundos. Às vezes as crises são representadas apenas por flexão da cabeça (tique de sabam ou "espasmo saudatório"). As crises são freqüentes particularmente durante a vigília, podendo chegar até a centena ou mais por dia.
As contrações são breves, maciças, simétricas, levando-se os membros superiores para frente e para fora e flexionando os músculos o abdômen. São crianças hipotônicas. Em princípio, o diagnóstico não é fácil, sendo os espasmos confundidos com cólicas ou com reflexo de Moro. Outra manifestação importante é o retardo mental que, em boa parcela dos casos, pode ser evitado pelo tratamento precoce do quadro. Diz-se que as alterações e características clínicas e evolutivas desta síndrome dependem das condições prévias do SNC do lactante antes dos surgimentos das crises. Com a maturação da criança , em geral as crises diminuem e desaparecem por volta do quarto ou quinto ano de vida.
Evolução, complicação e prognóstico
Quase sempre é uma perda de cunho neuropsíquico para criança afetada, esta perda está na dependência da precocidade de diagnóstico e da intervenção aplicada. A hipsarritmia pode desaparecer ou se transformar no decorrer do tempo. A criança apresenta sérias complicações respiratórias, devido aos freqüentes espasmos, deformidades, principalmente de MMSS e MMII. Pode ocorrer subluxação do quadril.
Há possibilidade de remissão total de espasmos infantis considerados criptogéticos, mas não há confirmação científica de remissão definitiva para os casos mais graves associados a outras condições ou patologias neurológicas.
Crianças apresentam sinais e sintomas de dano cerebral podem vir a apresentar um quadro de déficit intelectual a posterior, elas devem ser precocemente estimuladas para diminuir a seu grau de compartimento intelectual e psíquico.
Têm sido assinalados os casos em que o desenvolvimento é normal. Vários autores têm discutido uma associação entre a hipsarritmia e psicose ou entre hipsarritmia e autismo. A deteriorização do desenvolvimento neuropsicomotor está presente em 95 % dos casos. O melhor prognóstico ocorre nos 5% dos casos que permanecem com desenvolvimento mental.
O prognóstico, mesmo nos casos tratados precocemente, permanece reservado, observando-se em 90% dos casos a presença de deficiência mental. Distúrbios psiquiátricos são freqüentes. Outras síndromes epiléticas podem surgir, sendo que 50-60% dos casos evoluem para síndrome Lennox-Gastaut, epilepsia multifocal ou epilepsia parcial secundariamente generalizada.
Tratamento Clínico
Há uma grande melhora dos espasmos infantis com uso intensivo do ACTH (hormônio adrenocorticotrófico ) em suas apresentações injetáveis como: ACTHAR ( Corticotrophin) ou a sua forma de H.P.ACTHAR Gel (Repository Corticotrophin injection).
Dizemos que este tratamento pode ser heróico e interromper o quadro convulsivo, porém só deve ser utilizado sob rigoroso controle médico e monitoramento cardiopediatrico, já que os corticóides não agem apenas no SNC, mas todo o organismo da criança, inclusive em seu sistema imunológico. Segundo os autores Zajerman e Medina somente se utiliza esta medicação em casos de Síndrome de West considerados criptogênicos, e não em espasmos infantis resultantes de lesões cerebrais, por exemplo. Há casos em que a resposta terapêutica pode aparecer em até 48 ou 72 horas após aplicação de uma primeira dose do ACTH, podendo haver uma possibilidade de recidiva de crises nos casos considerados mais graves na dependência da precocidade do diagnóstico e da extensão e gravidade da lesão cerebral associada.
Outros anticonvulsivantes têm sido utilizados, isoladamente ou em combinação nos casos de espasmo infantis, como o Clonazepam, o Ácido Valpróico, o Fenobarbital e o Vigabatrin.
Tratamento Fisioterápico
Objetivos:
Equilíbrio da cabeça
Equilíbrio do tronco
Seguir as etapas de maturação de acordo com cada criança.
Em todo paciente com Síndrome de West precisa-se trabalhar primeiramente extensão de cabeça e de tronco, para que depois, então a criança seja estimulada a começar a rolar, arrastar, engatinhar, sentar, .. .Não podemos querer que ela engatinhe, sem que ela consiga fazer extensão cervical. O tratamento deve ser feito seguindo as etapas de evolução, de maturação da criança.
Os exercícios fisioterápicos devem obedecer as escalas de maturação. Tendo isso em mente, o fisioterapeuta pode inovar e criar novas maneiras de serem realizados de duas formas:
Utilizando a bola coloca-se a criança em DV apoiado com cotovelo em cima da bola, e chama-se a atenção da mesma com um objeto a sua frente.
Deitado no chão, também com um brinquedo à frente.
É importante saber que o tratamento da síndrome de west é igual ao tratamento proposto a criança portadora de paralisia cerebral.
Hidroterapia
Durante a terapia em piscina, o calor da água ajuda a aliviar a espasticidade, mesmo que o alívio seja temporário. Entretanto, a medida que a espasticidade diminui, movimentos passivos podem ser administrados em maior amplitude com menor desconforto para o paciente. Deste modo a amplitude articular pode ser mantida.
Os movimentos passivos devem ser efetuados lentamente e ritmicamente , começando com o tronco e articulações proximais, gradualmente incluindo as articulações distais. Os movimentos devem a princípio ser de natureza oscilatória e a seguir de natureza rotatória. O tronco e os membros devem ser movidos em padrões de movimento com inibição reflexa. O paciente deve respirar profundamente e calmamente, e o momento do estendimento máximo deve coincidir com a expiração. A principal dificuldade em obter uma fixação estável para ambos os pacientes e o terapeuta. Em alguns casos pode ser necessário um segundo fisioterapeuta para ajudar.
Tratamento fisioterápico
O tratamento fisioterapêutico tem como objetivo principal tratar as seqüelas ou tentar diminuí-las o máximo possível. Como as complicações respiratórias existentes, deve-se fazer fisioterapia respiratória.
Outro objetivo é tentar-se evitar as deformidades que surgem ou amenizá-las, fazendo-se mobilização passiva e alongamentos. Devido a hipotonia é preciso que se fortaleça os músculos responsáveis pela respiração.
REFERÊNCIA BIBLIOGRÁFICA:
http://www.coladaweb.com/biologia/genetica/sindromes-geneticas/sindrome-de-west
POSTADO POR: ARTUR FERREIRA COUTO
Nenhum comentário:
Postar um comentário